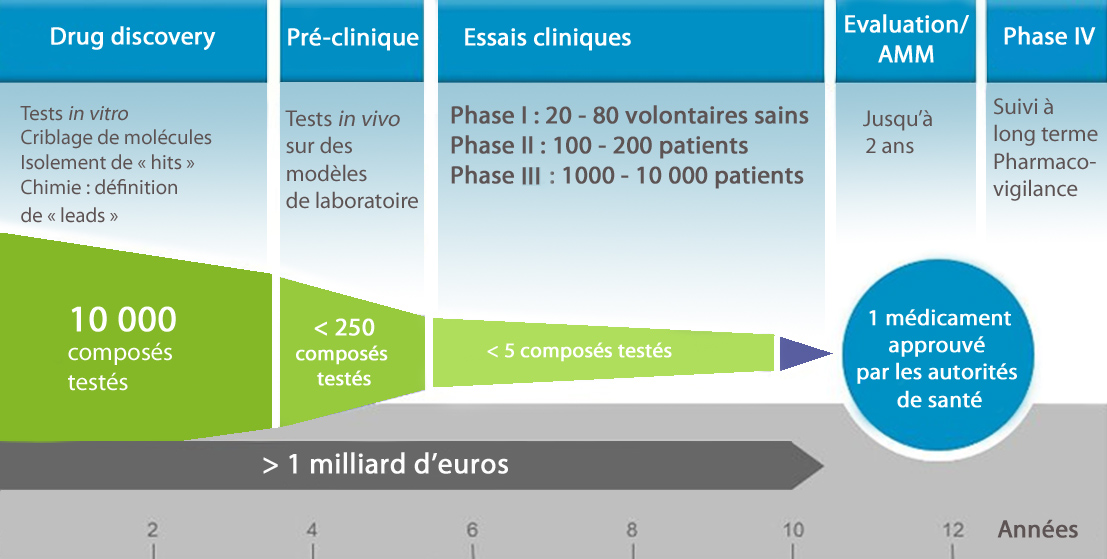
La recherche fondamentale représente le point de départ indispensable au développement de tout médicament. Elle se consacre à comprendre comment l’organisme fonctionne, de manière physiologique (normale) et pathologique (lors d’une maladie), ce qui permet de cerner finement comment une maladie se développe et donc, comment il est possible de la prévenir, la soulager, la soigner ou la guérir.
Dans le cas du cancer, les chercheurs essaient par exemple de comprendre comment une cellule normale devient cancéreuse, comment elle échappe au système immunitaire, comment elle forme une tumeur, comment celle-ci croît dans l’organisme, etc.
Dès lors que certains processus génétiques, cellulaires et/ou métaboliques anormaux particuliers sont mis en évidence, ils peuvent devenir des « cibles thérapeutiques ». L’enjeu est de trouver la bonne « flèche », c’est-à-dire une molécule chimique capable d’agir sur ces cibles (protéine, récepteur cellulaire, enzyme, gène…) pour entraver le développement cancéreux.
Il est aujourd'hui admis qu'en cancérologie, la recherche fondamentale est « le socle de toutes les innovations ».
La recherche fondamentale est habituellement réalisée dans des laboratoires universitaires publics.